Acute Neuronal and Systemic Inflammation caused by
Lipopolysaccharides of Salmonella minnesota can be
stopped by treatment with the Antibiotic Ofloxacin
Ines Niehaus
(contact: Ines_Niehaus@gmx.de)
Poster Presentation at the 6th Conference of the International Endotoxin Society
Abstract
In 1995 a female laboratory worker was contaminated with 10 microgram Salmonella minnesota S-LPS (lipopolysaccharides) through an open wound. One hour later she showed first symptoms of a strong LPS-induced inflammatory reaction (fever, flu-like-symptoms, cough with dyspnoea, headaches, nausea, vomiting). Three weeks after the contamination with LPS she experienced neurological problems (generalized cramps, disturbances of sensibility, myalgia, tremor, bradykinesia, parkinsonism, difficulties in learning etc.). The LPS damaged and disrupted the blood-brain barrier causing damages to the cerebral cortex and dopaminergic strial system proven by positron emission tomographies of the brain. The LPS, intercalating in cell and neuronal membranes, is eliciting a chronic systemic and neuronal inflammation. In 1999 a treatment with the antibiotic drug ofloxacin was started. Ofloxacin is binding to LPS and supposed to release LPS from cell and neuronal membranes. The patient was feeling quite better after a treatment with 200 mg ofloxacin for five days. But in 2000 the inflammation of the CNS turns back very heavily. In 2001 6600 pg LPS / ml cerebrospinal fluid were measured after one single dose of ofloxacin ten hours before the spinal tap. The chronic inflammation is still getting acute again once to two times a year. Ofloxacin is still able to reduce or to stop the acute LPS-induced inflammatory reaction but unable to cure the chronic inflammatory reaction to LPS. After stop taking ofloxacin LPS is sticking again to the cell and neuronal membranes. This is due to the fact that the S-LPS from Salmonella minnesota seems to be unable to be detoxified in this case report.
Introduction
Bacterial endotoxins (lipopolysaccharides) are part of the outer cell wall of Gram negative bacteria.1 Lipopolysaccharides (LPS) elicit multiple acute pathophysiological effects such as fever, lethality, Shwartzman reactivitiy, macrophage and B-lymphocyte activation, and other activities by animals and humans. 2, 3,4, 5LPS are highly biohazardous. Small doses of LPS (ca. 100 ng E. coli LPS) injected intravenously in humane volunteers are inducing short-lasting sepsis-like symptoms and activating important inflammatory mediators whereas large doses of LPS precipitating life-threatening circulatory collapse and multiple organ failure.6 1 mg Salmonella minnesota S-LPS leads to lethal septic shock in human beings without medical treatment.7
Gram-negative bacterial infections and toxins (e.g. LPS) have recently been implicated in the pathogenesis of a number of peripheral neuropathies in humans, including postinfectious polyneuritis or Guillain-Barre syndrome (GBS) and critical illness polyneuropathy (CIP). Encephalopathy and CIP occur in 70% of septic patients.10 Tumor necrosis factor and interleukin-1 (IL-1) and IL-2 are important mediators in the septic syndrome.10 The blood-brain barrier (BBB) becomes leaky in a patchy manner within the first few hours of sepsis.10I.p. injection of LPS at a dose below those described to disrupt the BBB, cytokine mRNA expression was increased in the spleen, the pituitary, and the brain.11 Disruption of the BBB can occur during a variety of pathophysiologic conditions, including bacterial infections. LPS increases the permeability of the BBB.12
LPS was quantified in plasma and CSF collected simultaneously from patients with systemic menigococcal disease.28 High levels (median 3800 ng/L; range, 750-14,000) were present in plasma and low levels (median, 40 ng/L; range, 25-165) in CSF of patients with septicemia. Conversely, high levels (median, 2500 ng/L; range, 25-500,000) in CSF and low or undetectabel levels (median, 25 ng/L; range, 25-210) in plasma were associated with meningitis without septic shock.28
A treatment of Gram-negative infections by fluoroquinolones (ciprofloxacin, ofloxacin, sparfloxacin e.g.) releases LPS from the bacterial cell wall. Treatment of Gram-negative infections of the central nervous system (CNS) with fluoroquinolones is very effective because the quinolones enter the cerebrospinal fluid (CSF) better than do any other class of antimicrobial agents. 13
In humans, an acute inhalation of pure endotoxin induces a blood and lung inflammatory reaction in which neutrophils and macrophages are involved and clinical symptoms, including fever and shaking chills, a lung function response characterized by bronchoconstriction and change in the level of nonspecific bronchial hyperresponsiveness. The chronic exposure of endotoxin has been related to both the risk of developing nonatopic chronic obstructive pulmonary diseases and the severity of domestic asthma. The threshold value of the no-response to LPS acute exposure is lower than 0,5 µg.14
LPS and lipid A are amphiphilic molecules and therefore form aggregates in aqueous environments above the critical concentration (critical aggregate concentration, CAC), which depends on their hydrophobicity and on the particular primary chemical struture.15 The CAC of LPS should be in the order of 10 -10 M or below.25 Above this concentration, there is an equilibirum between free monomers and aggregates.24 The CAC, which determines the number of free monomers, is supposed to be influenced by the amount of sugars linked to the lipid A portion of LPS as well as by the number and distribution of charges.24 Thus, the CAC should be highest for S-form LPS of wild-type strains, with high amounts of sugars represented by the O-specific charin, decreasing with the amount of sugars in LPS from rough mutant and deep-rough mutant strains, and be lowest for free lipid A. 24
A CD-14-independent, LBP-mediated transport of endotoxin molecules from aggregates directly into phospholipid membranes could recently be elucidated. Thus, LBP might act as a shuttle, causing a disaggregation of endotoxin aggregates by transporting the molecules directly or indirectly to target cell membranes. The LBP-mediated transport of endotoxin into liposomal phospholipid membranes corresponding to the composition of the macrophage cytoplasmic membrane. In the absence of serum proteins, the spontaneous intercalation of monomers into the macrophage membrane is likely to be part of the acivation process. The extent of this intercalation should be proportional to the number of monomers available, which increases with increasing length of the hydrophilic LPS region (higher CAC). The IL-6-inducing capacity increases in the sequence phosphate-free lipid A < dephosphorylated LPS Re and lipid A from LPS Re < LPS Re< LPS S-form. Endotoxin monomers, regardless whether existing per se or being liberated from aggregates by the action of LBP, may interact with the various endotoxin-binding proteins or may intercalate into host cell membranes, thus causing a further release of monomers from the aggregates to maintain the equilibrium, and thereby keeping the process in progress.24
Mean concentration of LBP was 1.72 µg/ml in normal CSF, i.e. 1/10 of the concentration measured in normal plasma. LBP concentration was found to be increased in CSF of patients with neurological disorders associated with increased protein levels in CSF between 2-fold and 7-fold, in patients with meningitis/encephalitis and with compressing tumors.27
The molecular shape of a given amphiphilic molecule like LPS or lipid A within a supramolecular aggregate depends, among others, on the fluidity (inversely correlated to the state of order) of its acyl chains, which can assume two phase states, the gel phase and the liquid-crystalline phase. Between these two phase states a reversible transition takes place at a given phase transition temperature TC, which depends, in the first place, on the length and the degreee of saturation of the acyl chains and on the conformation and the charge density of and its distribution within the headgroup region.15
At the physiologic temperature of 37°C, deep rough mutant LPS (Re LPS) exists in the liquid-crystalline fluid phase. Wild-type LPS, the natural form of LPS, consists of a mixture of different types of LPS that differ from each other in the length of the sugar moiety and in the acylation pattern of their hydrocarbon chains. Its TC is typically located around 37°C, and therefore its fluidity at 37°C is lower than that of Re LPS.26
LPS with longer sugar chains (e. g. S-LPS) and capsular polysaccharides are discussed as a physical barrier against terminal complement complex insertion.15Smooth (S) antigens probably allow resistance to phagocytes, since rough mutants (R) are more readily engulfed and destroyed by phagocytes. Smooth strains have polysaccharide "whiskers" which bear O antigens projecting from the cell surface. The O antigens are the key targets for the action of host antibody and complement, but when the reaction takes place at the tips of the polysaccharide chains, a significant distance external to the general bacterial cell surface, complement fails to have its normal lytic effect. Such bacteria are virulent because of this resistance to immune forces of the host. Rough strains are non virulent.16
Case Report
Ines Niehaus reports about her own case report:
In 1994/1995 at the age of 22 I was educated as a biologically technical assistant. Accidentally, I often breathed in LPS during my work in the lab regarding a time period about half a year. I had a permanent cough, chest pain and sometimes a light increase of temperature in the evening. I lost 5 kg weight during the half year. I am still suffering from endogenous asthma.
In March 1995 my left hand was injured (a scratch 1-2 cm long). 1 ml aqueous solution (RPMI medium for cell culturing) with 10 microgram Salmonella minnesota S-LPS leaked out of a 1 ml Eppendorf cap over the wound of my hand. At this moment I noticed the small wound for the first time, because it was burning. I washed my hands and desinfected the wound. I don't know about the biohazardous and pyrogenic properties of LPS and continued my work in the lab. One hour later I showed first signs of illness (fever, flu-like-symptoms, dyspnoea, headache, nausea). Later, the environment of the wound was swollen, red and the wound was healing bad. I had water filled blisters on my hands. My skin was very dry, pale and scaly so that each touch was painful. I was very thirsty although I drank 6-7 l water the following days. In the morning my temperature was 38-38,5°C and in the evening 39-40°C. I could not sleep because of the permanent cough, cold and dyspnoea. I was unable to eat something because of the nausea and vomiting. I nearly was unable to leave the bed due to vertigo, circulatory disorders and general weakness. My kidneys und urinary tract burned like fire. My family doctor thought I was suffering from an urinary tract infection and an influenza. But he did not examinate my blood or anything else. Against the overacidifying in my body I solved a package of baking powder (sodium hydrogen carbonate) in water and drank it. It stopped the pain of my kidneys.
Three weeks after my accident I got neurological problems (convulsions, sensory disturbances, myalgia, tremor, difficulties in learning and seeing etc.). I went to hospital but the physicians nothing understanding about LPS did not treat me in spite of high temperature (39°C-40°C), generalized cramps, vomiting etc. because no viral or bacterial infection was found although I had chronic leucocytes in my urine sample up to now. The result of an x-ray of the chest were normal. The electrocardiography I showed cardiac arrhythmias with a pulse of 80 to 110 per minute. One week later I was afebrile and I was discharged from the internal department of the hospital. After loosing 6 kg of body weight during these four weeks my body weight was only 49 kg at a body heigth of 175 cm. The physicians thought it was due to the fact that I did not want to eat. But I was eating as much as I am able to keep in my stomache during the day but in the nights I was vomiting for several weeks.
4 weeks after my accident the acute inflammatory reaction was over but I was still suffering from neurological problems especially cramp attacks without disorder of consciousness nearly every night.and myocloni in the nights. At day time I had a permanent tremor, myalgia in the legs, coordination disturbances, hyperaesthesia, hyperreflexia (every noise, vibration or touch led to muscle cloni). I often have a headache and could not bear sun light or flashes. 6 weeks after my accident I went to a neurological department of a hospital. Parkinsonism was diagnosed and a start of treatment with L-dopa was helpfully against the cramps in nights but this diagnose was falsely changed into rest-less legs syndrome, stiff-man syndrome, and others. A treament with drugs for parkinsonism were started again 6 years later in 2001 up to now (2007). Once to two times a year mostly in spring and/or autemn times the chronic inflammation of my nervous system is getting acute again with increase of temperature and very heavy pains of my nerves and deterioration of my symptoms of parkinsonism. In the worst case I had great difficulties in seeing, hearing, speaking and walking due to inflammation induced akinetic crises of PD. In summer 1998 great damages of my cerebral cortex were shown by PET. Therefore I was forbidden to work in a laboratory any longer because any contact to chemicals is deteriorating my symptoms. In 1999 it was the first time that I received glucocorticoids and analgetics which were helpful against the pains for a few hours but could not stop the inflammation.
In July 1999 starting a therapy with ofloxacin I could suddenly see nearly as clear as before my accident after the first dose of 100 mg ofloxacin. The second dose of 200 mg ofloxacin elicited a hot pain shooting through my cervical. I had severe pain in the spinal column and spinal nerves, which was coming down from the cervical. I reduced the next dose to 100 mg ofloxacin which stopped the pain. The next two days I took the regularely dose of 200 mg ofloxacin twice a day. I went to hospital because I could not bear the pains of my inflamed CNS any longer. There the physicians removed the ofloxacin from me after threee days taking it.
The free LPS, which was released from the neuronal membranes in my brain by ofloxacin, is able to aggregate to supramolecular structures in the cerebrospinal fluid. Ofloxacin is supposed to prevent this clustering by binding monomeric LPS molecules. In sitting or standing position of my body these multimeric LPS clusters could be stored in the lower parts of my spinal cord. I think I was able to feel it because of the heavy pains in this region of my spinal cord. In lying position these LPS aggregates were swimming in the CSF being able to rise in the spinal cord to the brain again. In the brain they are causing cramp attacks, which are stopping in sitting or standing position.
Half a year later the inflammation of the CNS turned back very heavily - it began with high temperature (+1°C-1,5°C) and heavy pains in the legs. I could stop this acute phase with glucocorticoids and analgetics at the beginning. On 03-03-2000 the pains of my legs and back were insufferable. A few single doses of 100 mg ofloxacin stopped this second acute phase. On 15-04-2000 the inflammation of the third acute phase reached my brain. I was unable to stop the fever or the heavy pains. On 16-04-2000 1:00 a. m. I took 400 mg ofloxacin. The fever increased up to 40,2°C. I was better for only 3 hours after taking 1200 mg ibuprofen. At daybreak I cried for pains and fever but a few days later I was better.
Since May/June 2000 I am better because the acute phase of inflammation was stopped but I am afraid of further phases of inflammation. Alas, acute phases of inflammation are/were coming back once to two times a year mostly in spring or autemn times also triggered by general exhaustion or secondary infections. I am unable to tolerate hot temperatures. Ofloxacin is still helpfully but it is unable to cure because the LPS of Salmonella minnesota are not being detoxified.
Materials and Methods
1) LAL test
Preparation of plasma for the limulus assay has been described in 28. CSF for the limulus assay was collected into LPS-free tubes without centrifugation and stored at -70°C. The chromogenic limulus lysate assay was used as described in 28.
2) PET
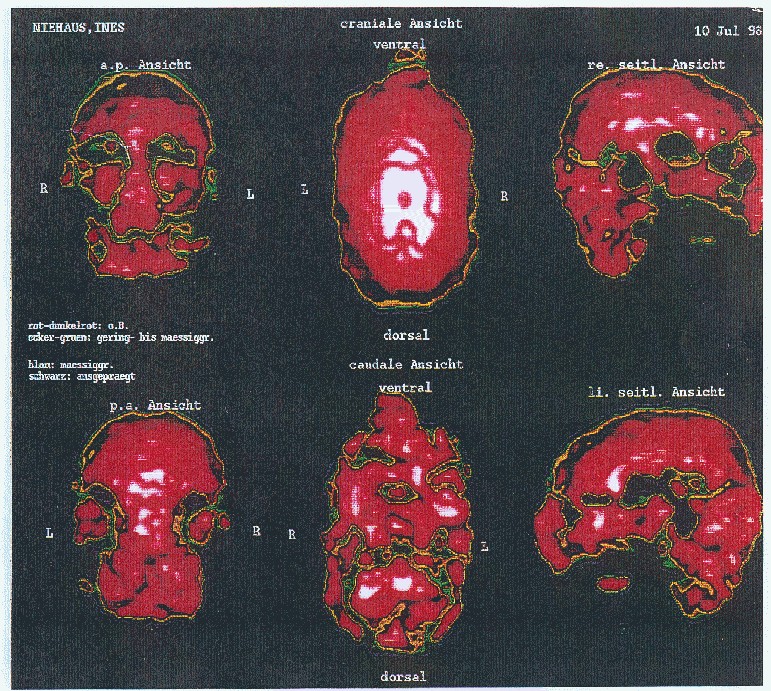
Cerebral glucose metabolism was determined with [Fluorine-18] fluoro-2-deoxy-D-glucose (FDG) using positron emission tomography (Fig. 1).
Under normal conditions, the human brain depends on glucose as its only energy source. The majority of glucose is used for maintaining the membrane potentials and restoring ion gradients. When clinically measuring glucose metabolism, the subject lies still for roughly 40 minutes after having FDG intravenously injected into their bloodstream. During this 40-minute time frame, the FDG is taken up by cells and will reach equilibrium. The distribution of the Fluorine-18 is then measured in multiple tomographic sections, and this distribution will be based on cerebral glucose utilization under the specific pathological or mental condition of the brain. 18
Figure 1. positron emission tomograph: http://www.alra.de
| Results
1) LAL test On 03.04.2000 the result of a LAL test of the CSF is 6600 pg LPS / ml. The result of a LAL test of the serum is negative.
|
2) Positron emission tomography with glucose (FDG)
patient: Ines Niehaus
born: 17.08.1972
date of examination: 10-07-1998
Table 1. reduction of glucose utilization in x% of the maximum activity of the cerebral cortex (100%)
| region | right side in % | left side in % |
| gyrus frontalis inferior | <70 | 70 |
| gyrus frontalis medius | <70 | 80 |
| gyrus frontalis superior | <70 | 80 |
| gyrus praecentralis | 75 | 80 |
| gyrus postcentralis | n.c. | 80 |
| lobulus parietalis inferior | 80 | 70 |
| gyrus temporalis inferior | 80 | n.c. |
| gyrus temporalis medius | 75 | 80 |
| gyrus temporalis superior | 70 | 80 |
| gyrus occipito-temporalis lateralis | 75 | <70 |
n.c.: no comment
Normally there are 100% activity in all regions of the cerebral cortex.
Figure 2a. Positron emission tomogram with FDG in the case report
annotation: red colour: normal; ochre-green: little damages; blue: moderate damages; black: strong damages
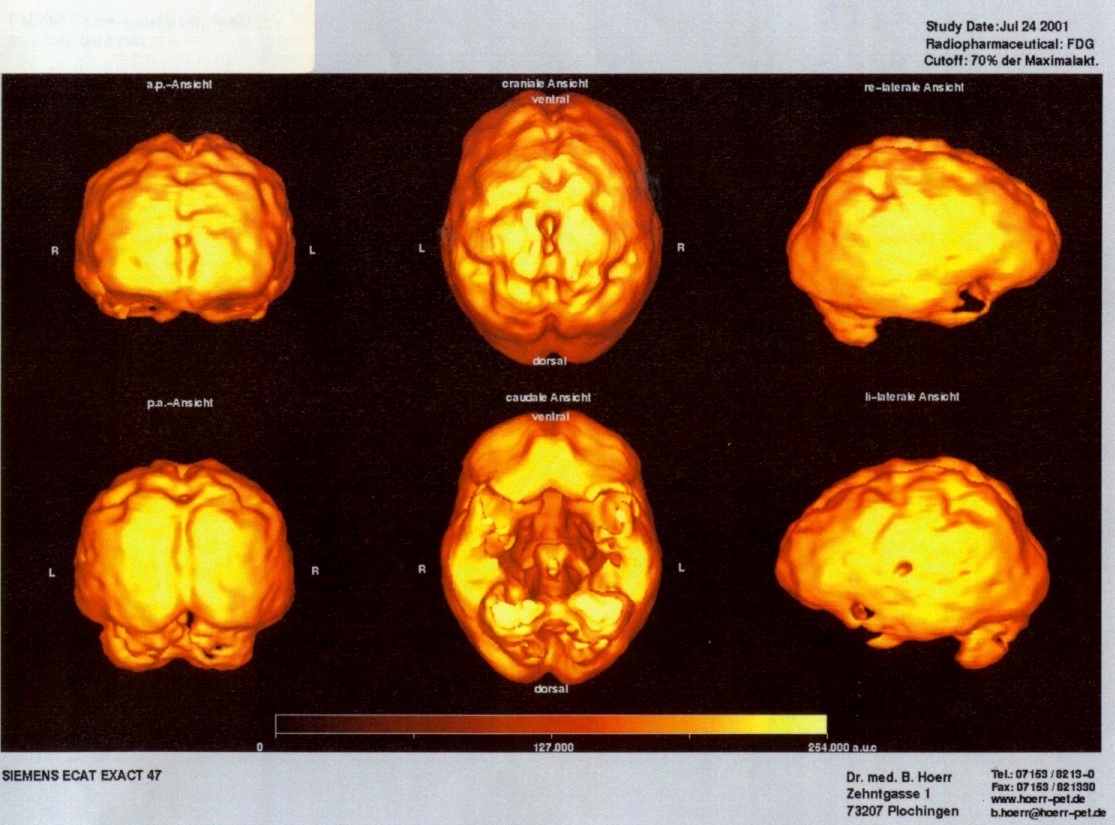
Figure 2b. Positron emission tomography with FDG in healthy volunteers for comparing with the PET of the patient
http://www.hoerr-pet.de/legende1.html (same PET-scanner)
Discussion
The dose of 10 µg Salmonella minnesota S-LPS I indicated as probable IV contamination is sufficient to cause some of the first symptoms (fever, flu-like-symptoms, dyspnoea, headache, nausea, vomiting). Robert Burell reports similar symptoms for doses of about 3-4 ng/kg which would be about 0.165µg for a 55 kg individual.3 Those doses were administred IV to healthy volunteers. LPS aggregates in aequous solution about the CAC (which was given by a solution of 10 µg / ml).15 Therefore it is probably that the total inoculation was 10 microgram LPS, which is equal to 182 ng per kilogram. Many humans are exposed each year to similar doses or higher during sepsis and septic shock. Encephalopathy and CIP occur in 70% of septic patients.24
Hyperreactivity to endotoxin may be caused by exotoxins, chronic infection, by growing tumors and probably like in this case by the contamination with LPS about the aerosol.19 I am suffering from endogenous asthma and bronchial hyperreactivity caused by the contamination with endotoxin about the aerosol. When the host organism is in a hyperreactive state to LPS low mediator concentrations may also become harmful.19 Moreover smooth (S) antigens probably allow resistance to phagocytes.16
Furthermore the contamination with LPS about the aerosol before the accident led to an allergic reaction on the S-LPS which was characterized by asthma (dyspnoea one and four hours after the accident), permanent cough during the first week after the accident (extrinsic allergic alveolitis?), blisters on the hands and heavy cold (rhinitis allergica?).
For that reason two reactions occured: the inflammatory and allergic response. That elicited a mild shock because of the disturbed water balance (I drank 5-6 l water a day), the circulatory disorders and the hyperacidity. I cannot prove it because medical examinations of any kind were not performed during the first three weeks after the accident. But a kind of shock is probable otherwise the endotoxin would not disrupt the BBB (see the positive LAL test of my CSF) and caused the damages of my cerebral cortex (see the PET) because endotoxin do not normally cross the BBB. My hypothesis is that the endotoxin has damaged and disrupted the BBB. The direct effect of cytokines released from macrophages on brain function may be enhanced by alterations in the BBB.10 Intracisternal or intraperitoneal injection of LPS produced disruption of the BBB to small moleculs (e.g. sodium fluorescein). 12 Topical application of LPS produced dilation of cerebral arterioles and increased the permeability of the BBB to FITC-dextran-10K.12 In contrast to FITC-dextran-10 K (molecular weight of 10000 Da) Lipid A from S.minnesota has only a molecular weight of 2036 Da, Ra-LPS 4236 Da and S-LPS ca. 10000 Da.19
Figure 3. Chemical structure of Salmonella LPS 19

Abbreviations: Glc: D-glucose; Gal: D-galactose; GlcNAc: N-acetyl-D-glucosamine; Hep: heptose; Kdo: 2-keto-3-deoxyoctonic acid; Abe: Abequose; Man: D-Mannose; Rha: rhamnose; n: the number of repeating units may vary from 0 to approximately 50.
In the brain the LPS are intercalated in the neurons with the lipid A so that the long sugar moiety of the S-LPS stand out from the membrane of the neurons. Anti-LPS antibodies hence activated complement etc. eliciting a chronical inflammation of the CNS because the nervous system was unable to destroy the LPS sticking in the neurons. The reactions of the immune system against the LPS were getting stronger and stronger by the time which elicited a continuous worsening of the complaints because of the increasing brain damages. After 4.5 years this inflammation process of the CNS was stopped by the treatment with the quinolone ofloxacin for a while.
Perhaps the LPS would be even intercalated into the lipid layer of the cellular membrane of the neurons similar as into the lipid layer of the cellular membrane of macrophages. Nonspecific, CD14-independent intercalation of LPS into membrane systems can be mediated by LBP. LBP breaks down LPS aggregates, transports the smaller units to and inserts them into the phospholipid cell matrix.25
LBP would act as a disaggregation protein and would guide LPS to several targets. Ofloxacin breaks down the size of LPS aggregates and amplifies the effect of LBP.
The LBP-driven intercalation of endotoxin does not require CD14 or any other membrane-associated protein. In living cells it may lead either to LPS neutralization or cell activation. A prerequisite for the activation of host cells is the insertion of endotoxin molecules into their lipid matrix. This can be achieved in different ways, (1) via direct intercalation by hydrophobic interaction of a-priori existing endotoxin monomers which are present in sufficient numbers at least for those chemotypes with longer sugar moieties (e. g. like in this case S. minnesota S-LPS), (2) via the intercalation of monomers or smaller endotoxin aggregates which are produced by the disaggregating properties of LBP, or (3) via the mediating role of mCD14. The mere intercalation of endotoxin molecules into the lipid matrix will not be sufficient for activation. The activation of host cells is based on the association of LPS with the target cell, resulting in an interdigitation of the lipid A acyl residues into the cell membrane. 25
At this time as the LPS disrupted the BBB the concentation of LBP was high in my CSF. The origin of the LBP in the CSF and/or the mechanisms by which is enters the CSF are unknown. 27 It can be hypothesized that any change of the permeability of the BBB could allow the passage of LBP to CSF. 27 So LBP triggers the intercalation of LPS in the lipid matrix of my neurons. Neurons cannot be activated by LPS and donot neutralize LPS so that the LPS sticked fixed in the membrane until such time as I started my treatment with ofloxacin.
Figure 4. The putative Mechanism of Intercalation of Salmonella minnesota S-LPS into Neuronal Membranes and the Ofloxacin-induced Release of LPS from the Neurons
Figure 4a. After disruption of the blood brain barriere the LPS are invading the neuronal extracellular space in the cerebrospinal fluid.
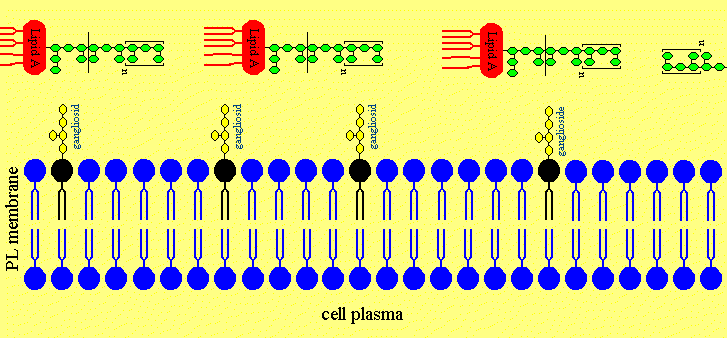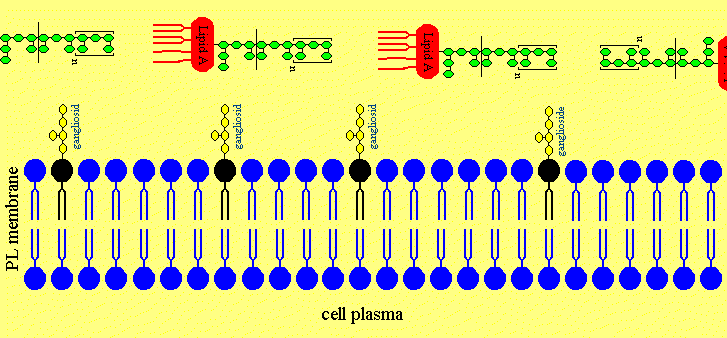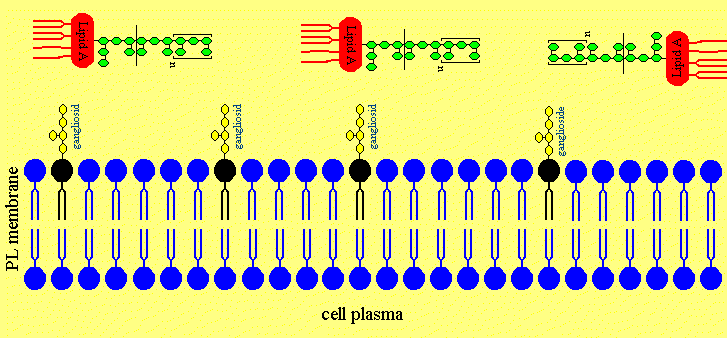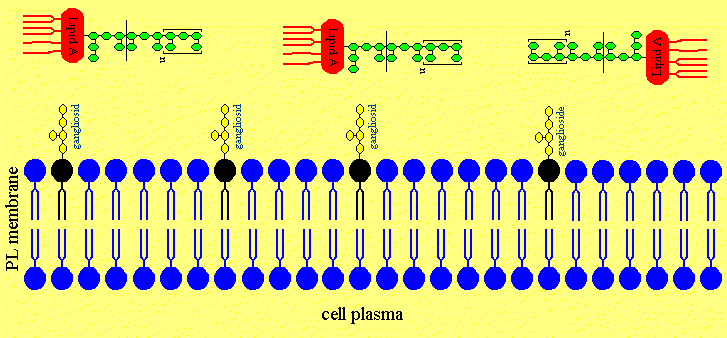
Figure 4b. The LPS is intercalating in neuronal membranes.
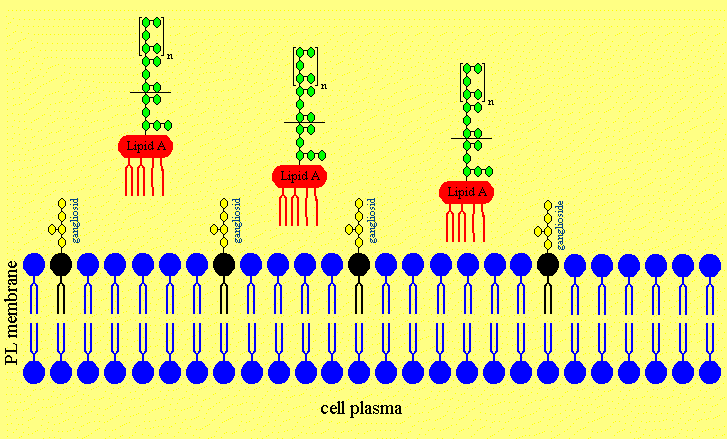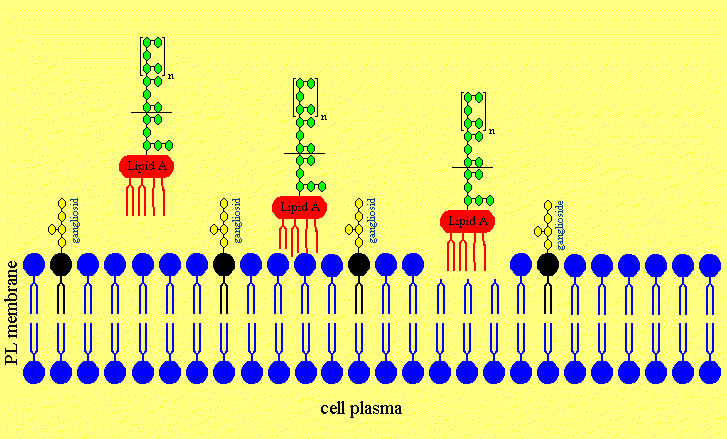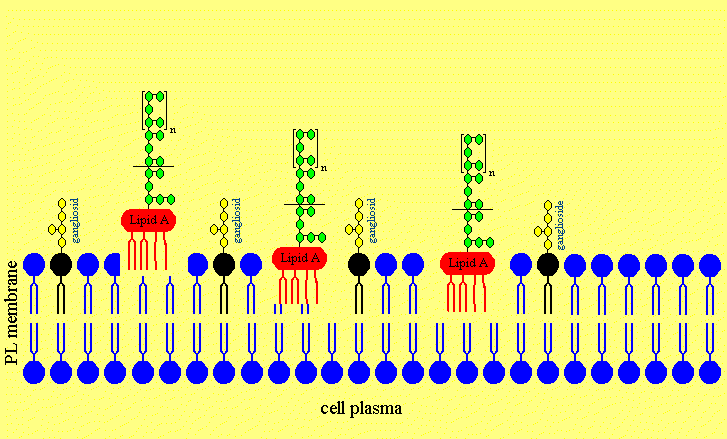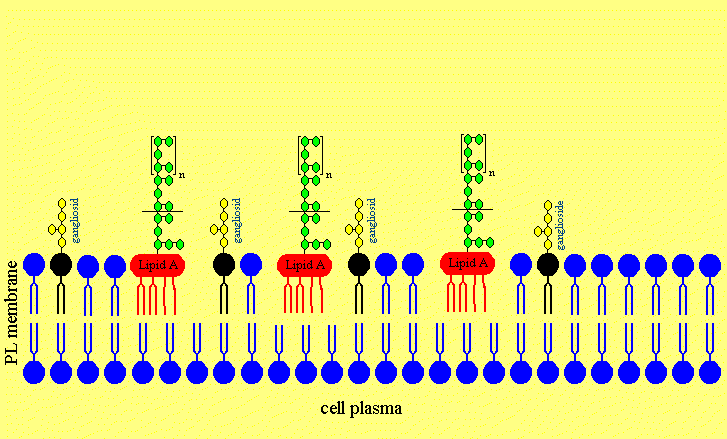
Figure 4c. The LPS is intercalated in the neuronal membrane and sticking fixed in the membrane with the Lipid A part of the LPS.
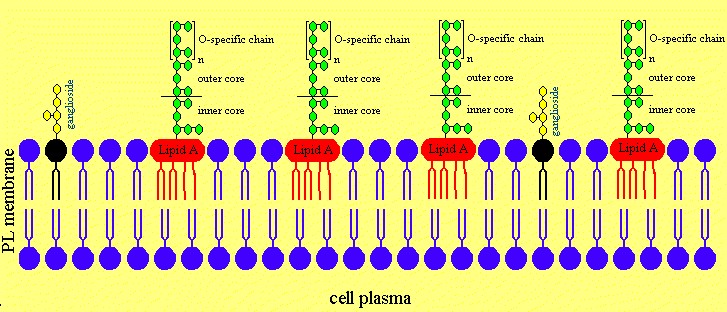
Figure 4d. Treatment with Ofloxacin leads to effective concentrations in the CNS.
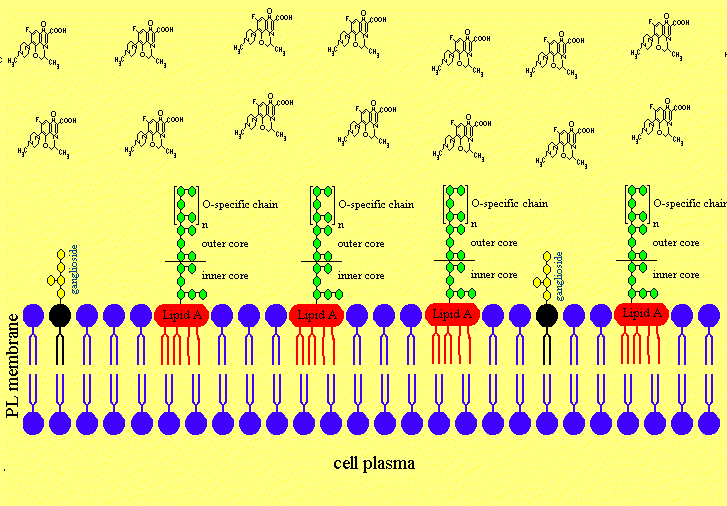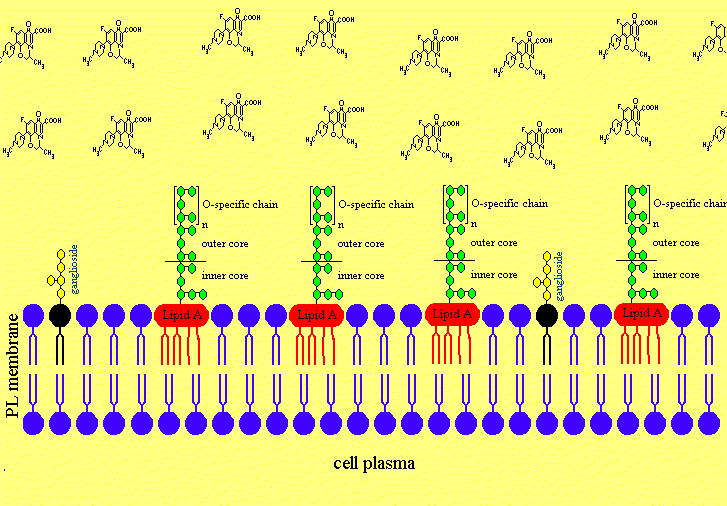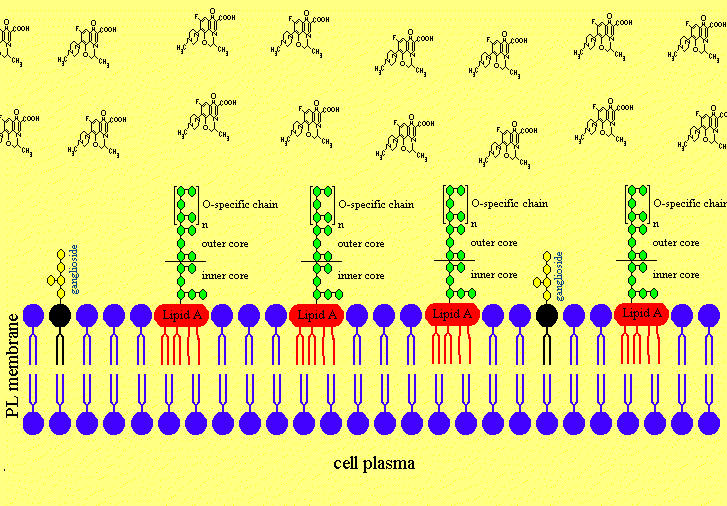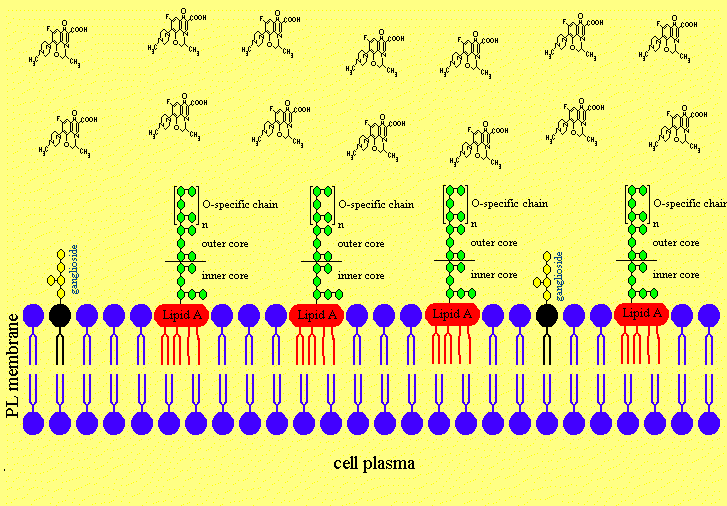
Figure 4e. Ofloxacin is releasing the lipopolysaccharides from neuronal membranes.
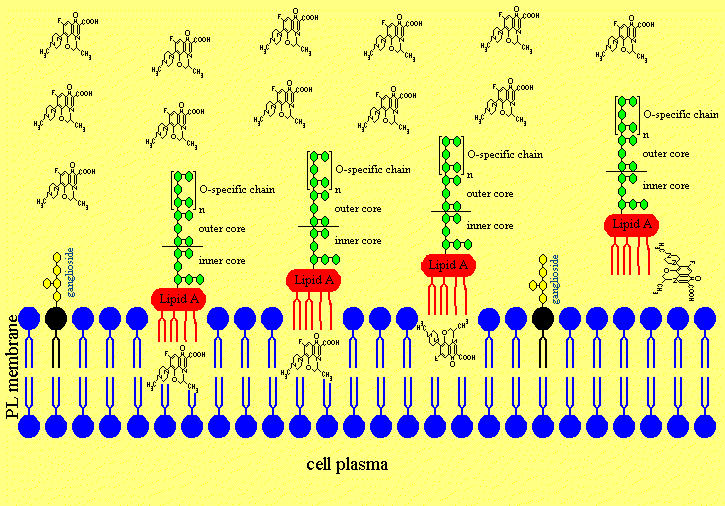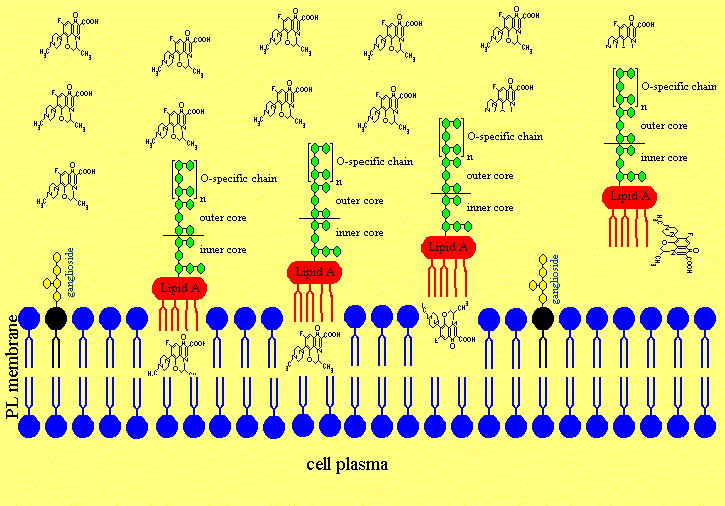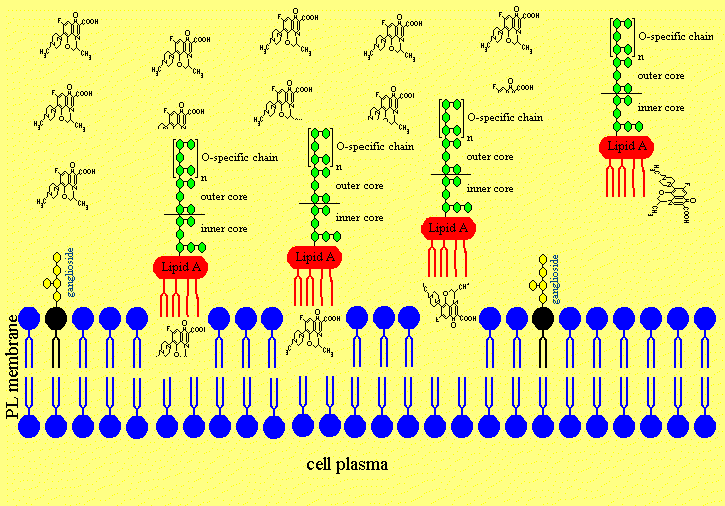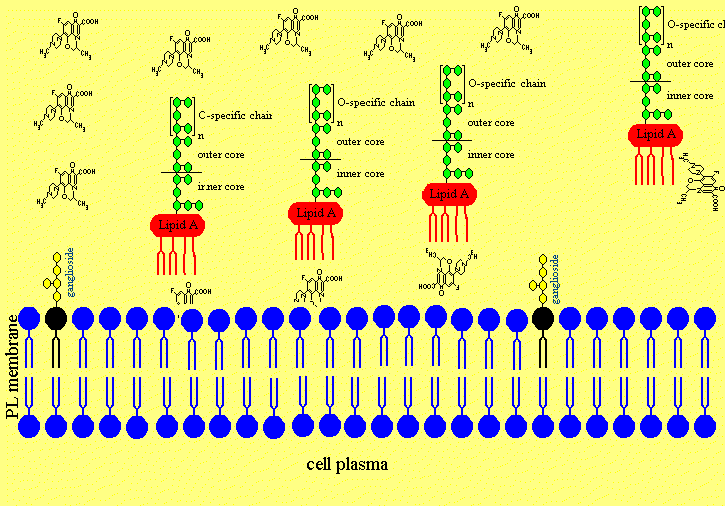
Figure 4f. Now the neuronal membranes are intact again.
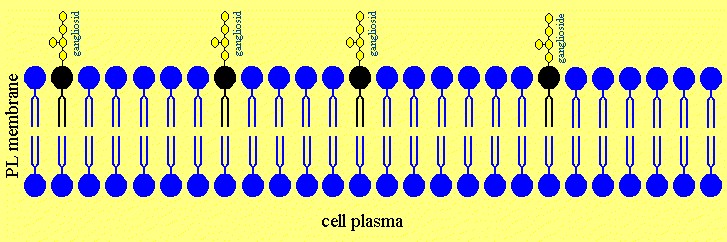
Ofloxacin is disturbing the order of the acyl chains of the Lipid A part of the LPS by hydrophobic interactions leading to an increased fluidity of the fatty acids of the lipid A. Therefore ofloxacin is able to release LPS of the cell and neuronal membrans similar as a treatment of Gram-negative infections with ofloxacin releases LPS from the bacterial cell wall. The LPS was sticking fixed in the membrans of cells and neurons but was released with the treatment of ofloxacin by causing a higher degree of fluidity of lipid A, which mobilised the lipid A part to removal from the phospholipid and ganglioside molecules of the cells.
Figure 5. Effect of Ofloxacin on Lipid A of Salmonella minnesota S-LPS. The numbers in circles represent the numbers of carbon atoms present in acyl chains of S-LPS of Salmonella minnesota.

During the therapy the CNS inflamed heavily caused by the free LPS in the CSF. The first dose of 100 mg ofloxacin was too low in order to reach the CSF in a effective concentration so that the better seeing was caused by a release of LPS from the neurons of the optic nerve. After the second dose of 200 mg the LPS releasing from the neurons in the brain could aggregate to supramolecular structures because the critical aggregation concentration is reached in this moment. After taking 100 mg ofloxacin the fluidity of the acyl chains of the lipid A was so enlarged by ofloxacin that these multimeric cluster could be destroyed.
The free LPS which was released from the neurons in my brain by ofloxacin were binding again to neurons after stop taking ofloxacin. The result of the LAL-test for my CSF was 6600 pg LPS / ml CSF in 2001.
At this time as the LPS was released from my neurons by ofloxacin I suppose that the level of LBP was normal because the BBB was intact. The low level of LBP was not able to disaggregate the big LPS cluster although LBP is functionally active in CSF. Ofloxacin broke down the LPS cluster in monomers in connection with the LBP so that the LBP could activate cells of monocytic origin in order to destroy the LPS. LBP is present in normal and inflamed CSF in concentrations that are suffucient to activate cells of monocytic origin. 27
So I think that ofloxacin might be amplifying the effect of LBP in CSF and perhaps in serum which has to be investigated.
One year later the result of a LAL test from my CSF is 6600 pg LPS/ml after taking 400 mg ofloxacin 10 hours before the spinal tap so that I did not destroy all LPS the year before. This concentration is with 6,6 x 10-10mol/l lower than the CAC. Therefore I did not have wandering LPS clusters in my CNS.
The hyperaesthesia, hyperreflexia and the sensory disturbances were caused by polyneuropathy. The putative mechanism is that LPS binds to peripheral nerve structures called gangliosides. This interaction is postulated to elicit the production of anti-LPS-antibodies, which then bind to the complex of LPS and gangliosides, and thereby induces neuropathological disease called GBS. GBS is a peripheral polyneuropathy, induced by infection with Campylobacter, and thought to be mediated by anti-ganglioside antibodies. GBS results from an immune response to cross-reactive antigen between LPS from Campylobacter jejuni and membran components of peripheral nerves, the gangliosides. 20 The same terminal tetrasaccharid occupies the nonreducing end of GM1 ganglioside and the LPS.20 This is called molecular mimicry between GM1 ganglioside and the LPS of C. jejuni.
Figure 6. Chemical structure of the ganglioside GM122

Abbreviations: Glc: glucose; Gal: galactose; NeuNAc: N-acetylneuraminic acid; GalNAc: N-acetylgalactosamine; blue: fatty acid (16, 18, 22 or 24 C-atoms); yellow: oligosaccharide; green: sphingosin
In contrast to Campylobacter jejuni LPS is Salmonella or E. coli LPS not similar to gangliosides (compare the yellow part of figure 3 and figure 6. But a case of GBS after Salmonella Ohio infection is known: the diagnosis of one patient presenting an acute weakness following an infection with Salmonella Ohio was GBS. However, preservation of reflexes and electrophysiological study showing purely axonal pattern make this case different from typical GBS. Outcome was favourable.21Anti-neuron antibodies and antibodies against specific gangliosides (group GM1) to prove a GBS like origin of the polyneuropathy were not found in my blood serum. So the LPS possible sticked in the neurons of the peripher nervous system and anti-LPS antibodies hence activated complement etc. Then after the treatment with ofloxacin I am suffering no longer from hyperaesthesia and hyperreflexia.
Conclusions
The sepsis was not treated after the contamination with 10 microgram Salmonella minnesota S-LPS through an open wound. The LPS disrupting the blood brain barriere is still intercalated in cell and neuronal membranes.
Ofloxacin is still helpful to reduce or to stop the acute phases of LPS-induced inflammations but is unable to cure. The LPS is sticking fixed again in the neurones after stop taking ofloxacin. Be aware that endotoxins, which are released from Gram negative bacteria during infections, are being detoxified in opposite to chemically highly purified LPS of Salmonella minnesota S-LPS in this case report. A future treatment option might be an extracorporeal removement of the LPS.
References
1) Rietschel ET, Brade H. Bacterial endotoxins. Sci Am 1992 Aug;267(2):54-61
2) Galanos C, Lüderitz O, Rietschel ET et al. Synthetic and natural Escherichia coli free lipid A express identical endotoxin activities. Eur. J. Biochem. 1985; 148(1): 1-5
3) Burrell R. Human responses to bacterial endotoxin. Circ. Shock 1994; 43(3): 137-153
4) Suffredini AF, Reda D, Banks SM, Tropea M, Agosti JM, Miller R. Effects of recombinant dimeric TNF receptor in human inflammatory responses following intravenous endotoxin administration. J Immunol 1995 Nov 15; 155(10): 5038-45
5) Pollmacher T, Mullington J, Korth C et al. Diurnal variations in the human host response to endotoxin. J Infect Dis 1996; 174(5): 1040-5
6) Brandtzaeg P. Significance and pathogenesis of septic shock. In: Rietschel ET, Wagner H, ed. pathology of septic shock, Springer-Verlag 1996:20-21
7) Taveira da Silva AM, Kaulbach HC, Chuidian FS, Lambert DR, Suffredini AF, Danner RF. Brief report: shock and multiple-organ dysfunction after self-administration of Salmonella endotoxin. N Engl J Med 1993; 328(20): 1457-60
8) Brown RF, Jackson GD, Martin T, Westbrook RF. Bacterial lipopolysaccharides induce peripheral nerve disturbances in rats that mimic human immune-mediated polyneuropathies. Lab Anim Sci 1997; 47(4): 354-61
9) Brown RF, Jackson GD, Martin T, Westbrook RF, Pollard JD, Westland KW. Bacterial lipopolysaccharide induces a conduction block in the sciatic nerves of rats. Lab Anim Sci 1999; 49(1): 62-69
10) Bolton CF, Young GB, Zochodne DW. The neurological complications of sepsis. Ann Neurol 1993; 33(1): 94-100
11) Pitossi F, del Rey A, Kabiersch A, Besedovsky H. Induction of cytokine transcripts in the central nervous system and pituitary following peripheral administration of endotoxin to mice. J Neurosci Res 1997; 48(4): 287-98
12) Mayhan WG. Effect of lipopolysaccharide on the permeability and reactivity of the cerebral microcirculation: role of inducible nitric oxide synthase. Brain Research 1998; 792(2): 353-357
13) Scheld WM. Quinolone therapy for infections of the central nervous system. Rev Infect Dis 1989; 11 Suppl 5: S1194-202
14) Michel O, Nagy AM, Schroeven M et al. Dose-response relationship to inhaled endotoxin in normal subjects. Am J Respir Crit Care Med 1997; 156(4 Pt 1): 1157-64
15) Seydel U, Wiese A, Schromm AB, Lindner B, Brandenburg K. Function and activity of endotoxins: a biophysical view. In: Forschungszentrum Borstel, Jahresforschungsbericht 1996: 7-52
16) Todar K. Bacteriology 330 lecture topics: bacterial endotoxins. Internet 1997
http://www.bact.wisc.edu/bact330/lectureendo
17) Sewerynek E, Melchiorri D, Chen L, Reiter RJ. Melatonin reduces both basal and bacterial lipopolysaccharide-induced lipid peroxidation in vitro. Free-Radic-Biol-Med. 1995; 19(6): 903-906
18) Mullen RJ. Positron Emission Tomography. Internet 1995
http://www.bae.ncsu.edu/bae/courses/bae590f/1995/mullen/
19) Rietschel ET, Kirikae T, Schade FU et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994; 8(2): 217-225
20) Yuki N, Taki T, Inagaki F et al. A bacterium lipopolysaccharide that elicits guillain-barre syndrome has a GM1 ganglioside-like structure J. Exp. Med. Nov 1993; 178(5): 1771-1775
21) Henriet M, Manto M, Jucquy J. Acute motor axonal neuropathy with favourable outcome after Salmonella Ohio infection. Rev Neurol (Paris) 1994; 150(10): 732-3
22) Lehninger AL, Nelson DL, Cox MM. Lipide. In: Prinzipien der Biochemie, second edition 1994: S. 279-309
23) Rietschel, Ernst Theodor: Bakterielle Endotoxine: chemische Konstitution und biologische Wirkung Nordrhein-Westfälische Akademie der Wissenschaften, 433. Sitzung am 3. Dezember 1997 in Düsseldorf (Vortrag)
24) Schromm AB, Brandenburg K, Loppnow H et al. The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J of Immunol 1998; 161:5464-5471
25) Schromm AB, Brandenburg K, Rietschel ET, Flad HD, Carroll SF, Seydel U. Lipopolysaccharide-binding protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS Lett 1996; 399: 267-271
26) Wellinghausen N, Schromm AB, Seydel U et al. Zinc enhances lipopolysaccharide-induced monokine secretion by alteration of fluidity state of lipopolysaccharide. J of Immunol 1996; 157: 3139-3145
27) Heumann D, Gallay P, Barras C et al. Lipolysaccharide binding protein (LBP) as a marker of protein laekage in cerebrospinal fluid. J of Endotoxin Research 1995; 2: 1-7
28) Brandtzaeg P, Kierulf P, Gaustad P et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis 1989; 159:195-204
This page is edited by Ines Niehaus on 18th October 1999.
last update: 8th November 2007
e-mail: Ines_Niehaus@gmx.de